





































































一、获取SRA号
登陆Genome Announcements网站(地址:https://mra.asm.org/),搜索感兴趣的基因组,我搜索的是细菌基因组。
进入网站页面后,点击右上角的搜索符号。
我随意选了一篇文章,是一篇菲律宾贝纳姆河岸分离的六种细菌基因组序列草稿的文章。我在文章里找到了细菌信息。
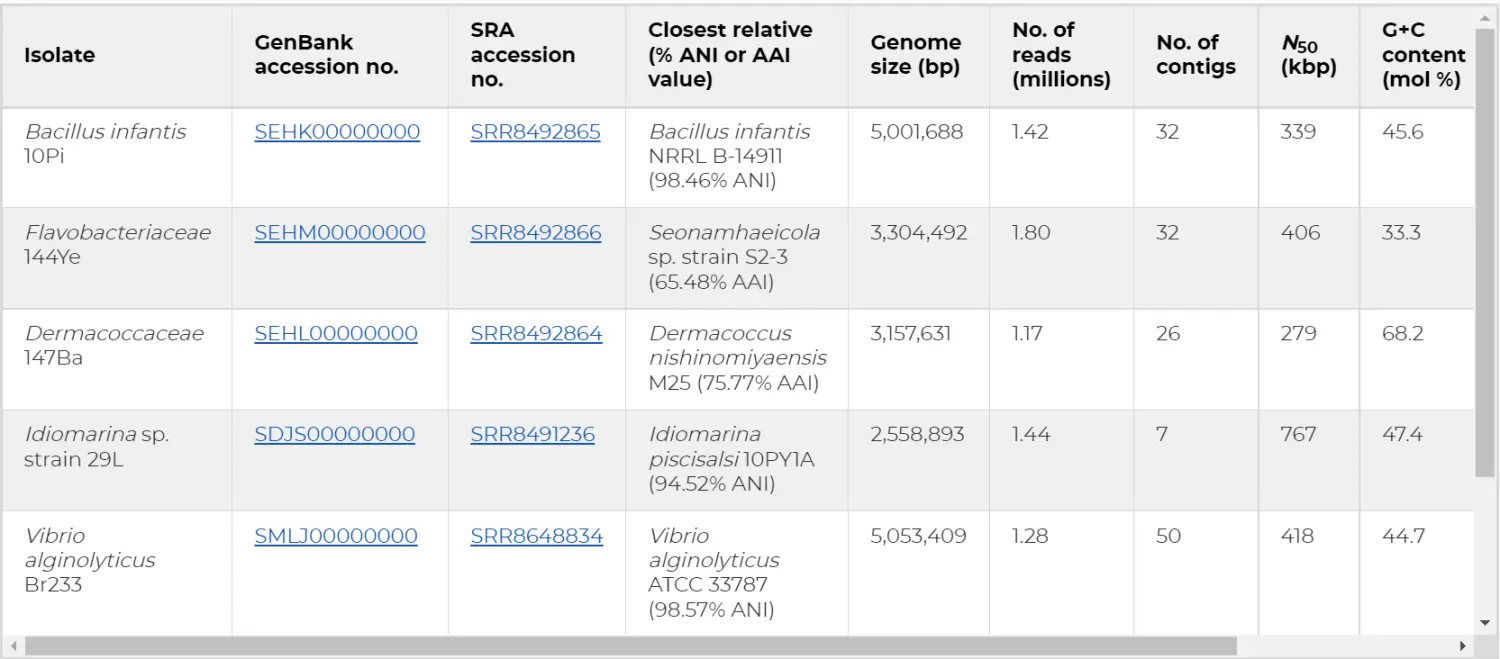
我选取第四个细菌基因组为例。
SRR8491236
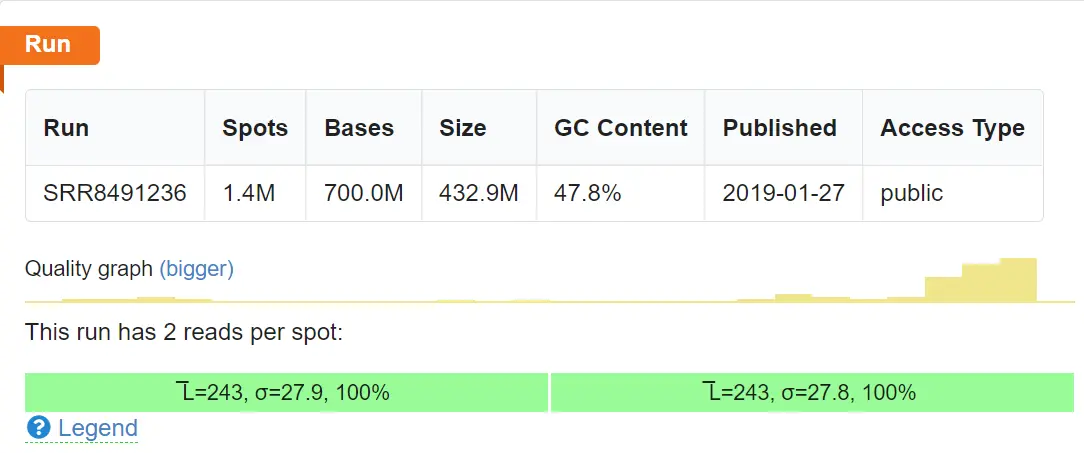
二、下载SRA序列
首先需要在linux下载SRAtoolkit,然后才能下载SRA数据。
可以参考我的简书文章:
https://www.jianshu.com/p/3df52aa857ad?v=1667697177169
然后再执行命令将SRA数据下载下来:
prefetch SRR8491236
运行结果如下:
一般软件会自动建立~/ncbi/public/sra文件夹,但我的不知什么缘由,经常不会建立这个文件夹,所以我就手动建立了。

三、解压SRA文件为fastq格式
命令如下:
fastq-dump --gzip --split-files SRR8491236
结果如下:
四、用fastqc进行数据质量评价
这里需要安装fastqc,可以参考我的简书文章:
https://www.jianshu.com/p/5d364dfc0f43
输入以下命令:
fastqc SRR8491236_1.fastq.gz
fastqc SRR8491236_2.fastq.gz
结果如下:
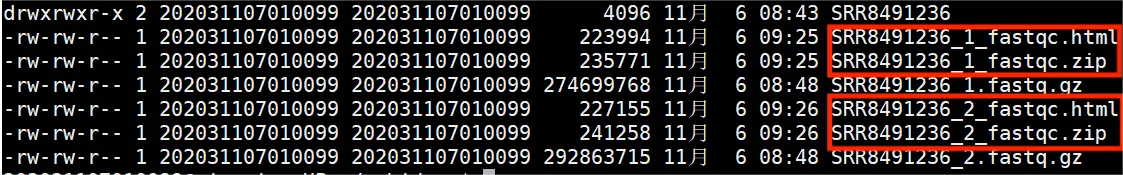
每个测序样本生成了两个文件。
可以将html文件下载到本地来查看和分析。
FastQC Report解读
FastqC有3种结果:绿色代表pass;黄色代表warn;红色代表fail。
可以参考简书文章:https://www.jianshu.com/p/c81c7110eed4
五、Trimmomatic去接头
Trimmomatic是一个广受欢迎的Illumina 平台数据过滤工具。
Trimmomatic支持多线程,处理数据速度快,主要用来去除Illumina 平台的Fastq序列中的接头,并根据碱基质量值对Fastq进行修剪。
软件有两种过滤模式,分别对应SE 和PE 测序数据,同时支持gzip和bzip2 压缩文件。
Trimmomatic也支持phred-33 和phred-64 格式互相转化,不过现在绝大部分Illumina 平台的产出数据也都转为使用phred-33 格式了。
Trimmomatic是基于Java开发的,因此需要提前安装Java,才能使用Trimmomatic。
Trimmomatic安装可以参考我的简书文章:
https://www.jianshu.com/p/8e854198db13
Trimmomatic过滤命令如下:
java -jar Trimmomatic-0.38/trimmomatic-0.38.jar PE -phred33 ~/ncbi/sra/SRR8491236_1.fastq.gz ./ncbi/sra/SRR8491236_2.fastq.gz ./output_forward_paired.fq.gz ./output_forward_unpaired.fq.gz ./output_reverse_paired.fq.gz ./output_reverse_unpaired.fq.gz ILLUMINACLIP:Trimmomatic-0.38/adapters/TruSeq2-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:20 TRAILING:20 MINLEN:75
命令执行解释:
PE -phred33 因为双端测序需要输入两个文件。最终会输出4个文件。其中两个文件对应于"paired"数据,即read1和read2都保留。另外两个对应于"unpaired"数据,在处理的过程中会过滤掉其中一端的reads。
~/ncbi/sra/SRR8491236_1.fastq.gz ./ncbi/sra/SRR8491236_2.fastq.gz 这里是输入将要过滤的文件
./output_forward_paired.fq.gz ./output_forward_unpaired.fq.gz ./output_reverse_paired.fq.gz ./output_reverse_unpaired.fq.gz这里是输出的四个结果文件,都保存在当前目录下
ILLUMINACLIP:Trimmomatic-0.38/adapters/TruSeq2-PE.fa:2:30:10 切除TruSeq3-PE中提供的Illumina适配器。最初Trimmomatic将寻找种子匹配(16个碱基),最多允许2个不匹配。如果在配对端读取的情况下达到30分(约50个碱基,50*0.6),或在单端读取的情况下达到10分(约17个碱基, 17*0.6),这些"seed"序列将被修剪。
SLIDINGWINDOW:5:20 扫描5个碱基宽滑动窗口,当每个碱基的平均质量下降到20以下时进行剪切
LEADING:20 删除前低质量碱基或N碱基(低于质量3)
TRAILING:20 删除后低质量碱基或N碱基(低于质量3)
MINLEN:75 上述步骤完成后,删除小于36个碱基的reads
参考文章:
https://www.cnblogs.com/Sunny-King/archive/2022/06/16/Bioinformatics-Trimmomatic.html
运行结果:
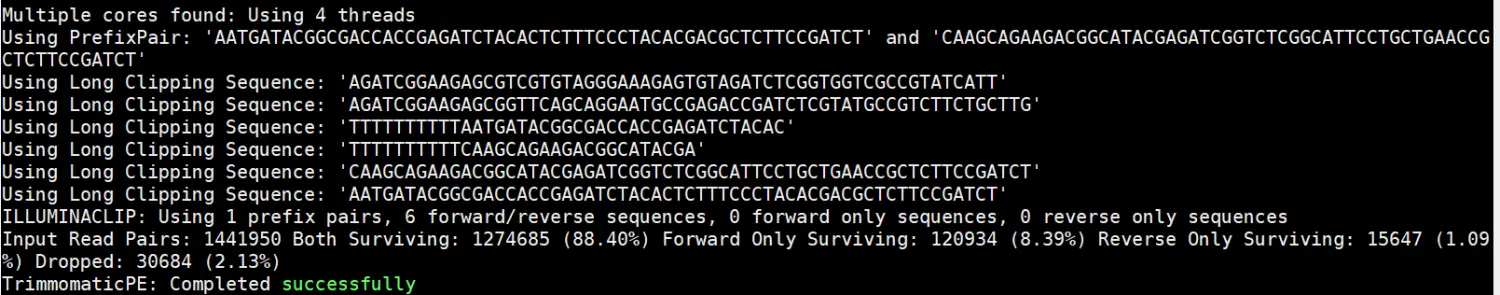
由结果可以知道,我的两条双端测序都保留的占88.40%,只有正向测序的序列保留的占8.39%,只有反向测序的序列保留的占1.09%,过滤掉2.13%。
六、Spades组装基因组草图
安装Spades可以参考我的简书文章:
https://www.jianshu.com/p/f5b2aa3fda64?v=1667780854519
运行命令如下:
spades.py --careful --pe1-1 ~/ncbi/sra/SRR8491236_1.fastq.gz --pe1-2 ~/ncbi/sra/SRR8491236_2.fastq.gz -o ./SPAdesout_SRR8491236
命令解释:
--careful 减少错配和短插入失.让程序运行MismatchCorrector----不建议用于中型和大型基因组。官方文档中可以看到MismatchCorrector占用的disk space非常多。
注:默认情况下执行read纠错、组装。
--pe1-1
--pe<#>-1 <file_name>:left reads, <#>代表第几个文库。
--pe<#>-2 <file_name>:right reads, <#>代表第几个文库。
note:只有一个文库,指定#等于1即可。
-o 指定输出文件目录(必需)。
这里参考简书文章:
https://www.jianshu.com/p/f02475d61a5c
出现错误,内存空间不足:
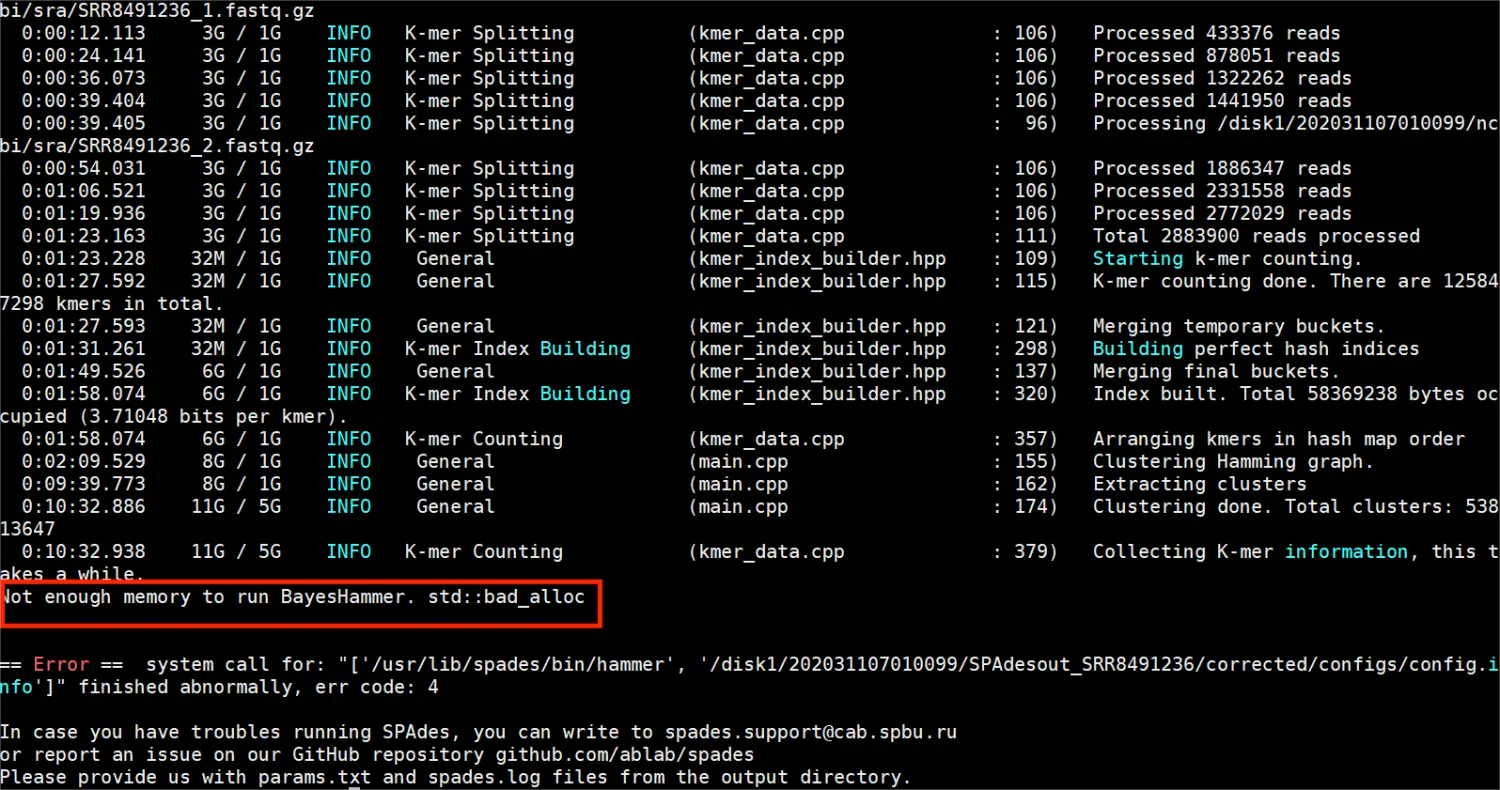
我尝试使用seqtk抽取1000条,命令如下:
#解压
gunzip -c output_forward_paired.fq.gz > output_forward_paired.fq
gunzip -c output_reverse_paired.fq.gz > output_reverse_paired.fq
#抽取1000条
seqtk sample -s60 output_forward_paired.fq 1000 > seqtksample1_1000.fq
seqtk sample -s60 output_reverse_paired.fq 1000 > seqtksample2_1000.fq
#用wc查看,可对比前后文件,判断是否抽取成功
wc -l output_forward_paired.fq
wc -l seqtksample1_1000.fq
spades.py --careful --pe1-1 seqtksample1_1000.fq --pe1-2 seqtksample2_1000.fq -o ./SPAdesout_SRR8491236_1000
这里需要下载seqtk。关于下载seqtk,可以参考我的简书文章:
https://www.jianshu.com/p/316b706a8bc9?v=1667780899292
命令解释:
gunzip -c #-c或--stdout或--to-stdout 把解压后的文件输出到标准输出设备。
seqtk sample -s60 从pair end的原始fastq文件中抽取1000条reads。其中-s是seed,控制随机抽取,但是要注意在抽R1和R2的时候,一定要用相同的seed,这样才能保证抽出来的R1和R2仍然是配对的,否则有可能会错位。后面1000表示抽取的reads数目。
wc -l 统计文件内容的行数
参考文章:https://cloud.tencent.com/developer/article/1674827
我还是出错了。
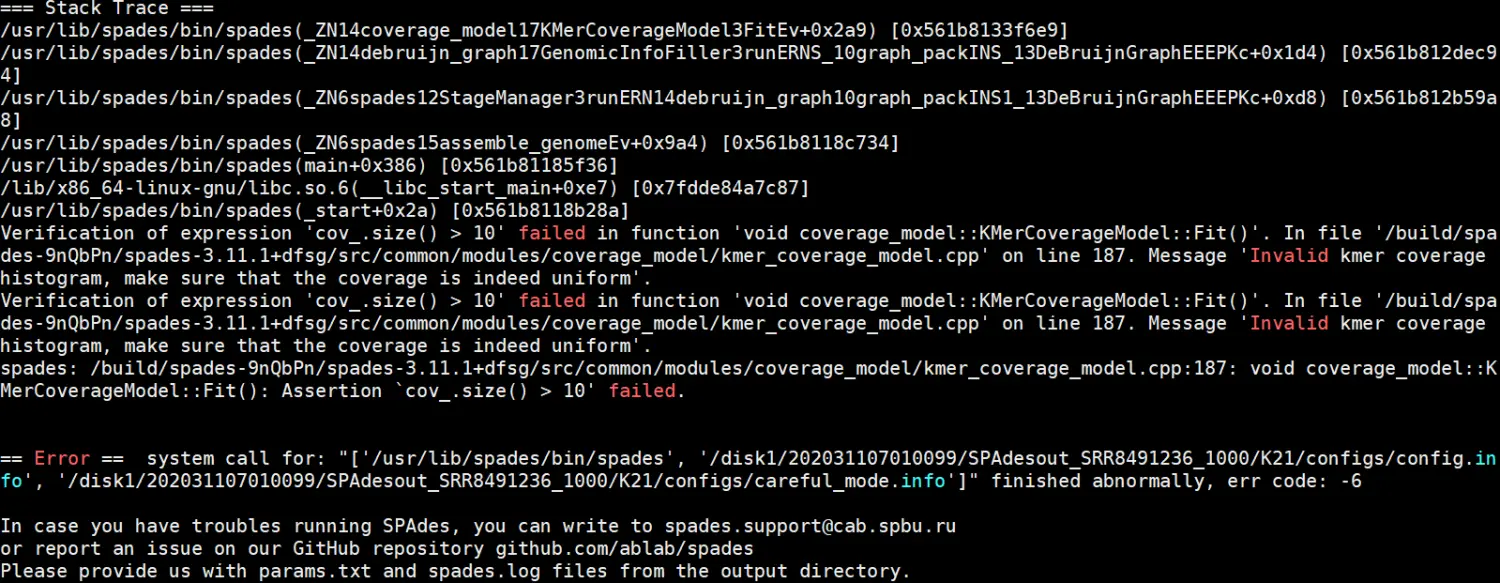
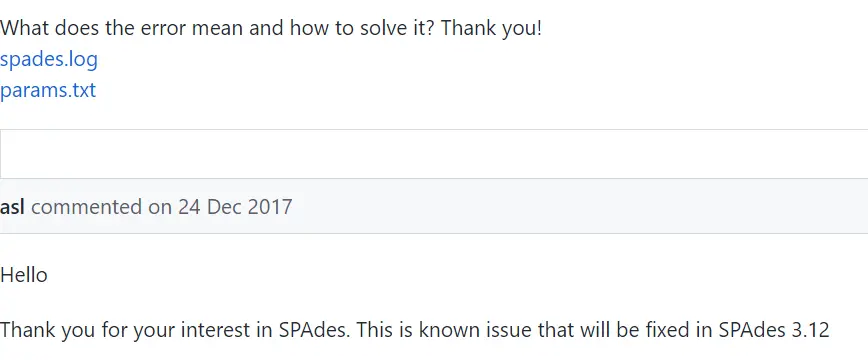
看到这个回答,我将SPAdes卸载了,安装了最新版。我原来安装的版本是3.12.0,现在安装的最新版本是3.15.4。
然后我重试,又出现了问题:
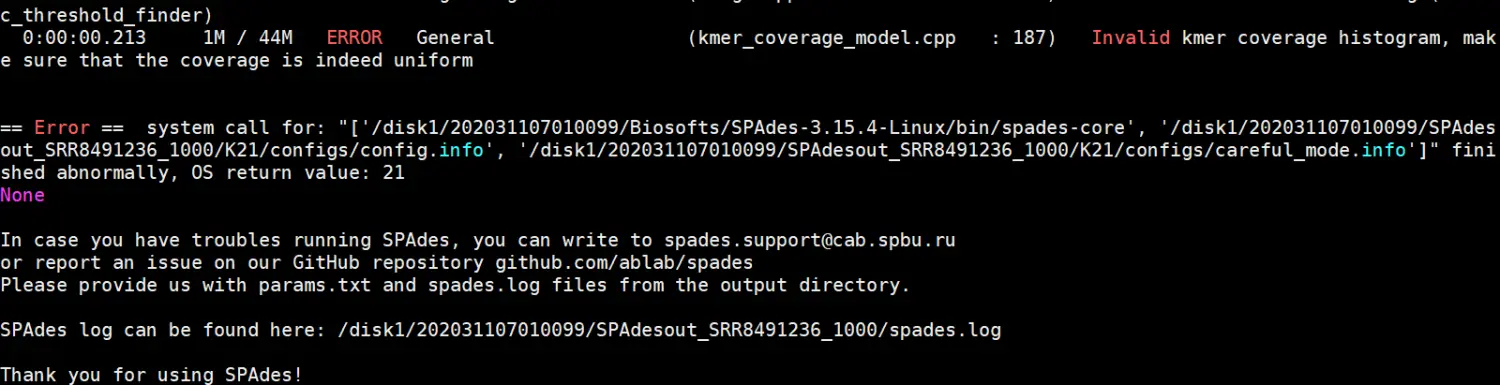

根据这个回答,我输入如下命令:
spades.py --sc --careful --pe1-1 seqtksample1_1000.fq --pe1-2 seqtksample2_1000.fq -o ./SPAdesout_SRR8491236_1000
结果如下:

感觉应该是成功了吧。
七、Quast评价组装的基因组效果
武汉格发信息技术有限公司,格发许可优化管理系统可以帮你评估贵公司软件许可的真实需求,再低成本合规性管理软件许可,帮助贵司提高软件投资回报率,为软件采购、使用提供科学决策依据。支持的软件有: CAD,CAE,PDM,PLM,Catia,Ugnx, AutoCAD, Pro/E, Solidworks 等。

 技术文档
技术文档
 推荐好文
推荐好文







 155-2731-8020
155-2731-8020