















































软件

产品


MS Discover 结构优化原理
分子的势能一般为键合(键长、键角、二面角、扭转角等)和非键合相互作用(静电作用、范德华作用等)能量项的加和,总势能是各类势能之和,如下式:


除了一些简单的分子以外,大多数的势能是分子中一些复杂形势的势能的组合。势能为分子中原子坐标的函数,由原子不同的坐标所得到的势能构成势能面(Potential Energy Surface,PES)。势能越低,构象越稳定,在系统中出现的机率越大;反之,势能越高,构象越不稳定,在系统中出现的机率越小。通常势能面可得到许多极小值的位置,其中对应于最低能量的点称为全局最小值(Global Energy Minimum),相当于分子最稳定的构象。由势能面求最低极小值的过程称为能量最小化(Energy Minimum),其所对应的结构为最优化结构(Optimized Structure),能量最小化过程,亦是结构优化的过程。
通过最小化算法进行结构优化时,应避免陷入局部最小值(local minimum),也就是避免仅得到某一构象附近的相对稳定的构象,而力求得到全局最小值,即实现全局优化。分子力学的最小化算法能较快进行能量优化,但它的局限性在于易陷入局部势阱,求得的往往是局部最小值,而要寻求全局最小值只能采用系统搜寻法或分子动力学法。
在Materials Studio的Discover模块中,能量最小化算法有以下四种:
1)最陡下降法(Steepest Descent),为一经典的方法,通过迭代求导,对多变量的非线性目标函数极小化,按能量梯度相反的方向对坐标添加一位移,即能量函数的负梯度方向是目标函数最陡下降的方向,所以称为最陡下降法。此法计算简单,速度快,但在极小值附近收敛性不够好,造成移动方向正交。最陡下降法适用于优化的最初阶段。
2)共轭梯度法(Conjugate Gradient),在求导时,目标函数下降方向不是仅选取最陡下降法所采用的能量函数的负梯度方向,而是选取两个共轭梯度方向,即前次迭代时的能量函数负梯度方向与当前迭代时的能量函数负梯度方向的线性组合。此法收敛性较好,但对分子起始结构要求较高,因此常与最陡下降法联合使用,先用最陡下降法优化,再用共轭梯度法优化至收敛。
3)牛顿方法(Newton),以二阶导数方法求得极小值。此法的收敛很迅速,也常与最陡下降法联合使用。
4)综合法(Smart Minimizer),该方法可以混合最陡下降法,共轭梯度法和牛顿法进行结构优化,在MS中是可选择的。
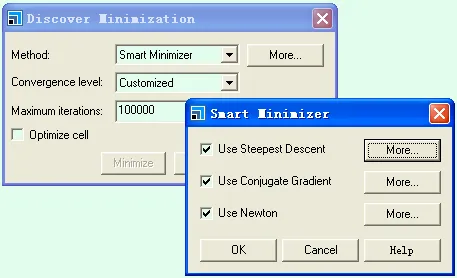
点开各种方法后面的More,可设定收敛精度(Convergence),算法(Algorithm)和一维搜索(Line search,指每一次迭代中的精度)等。
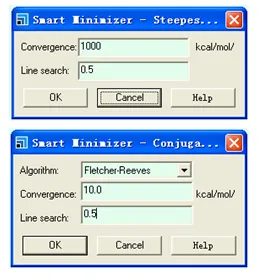
Smart Minimizer中,只有共轭梯度法和牛顿法才可以选择不同的算法:

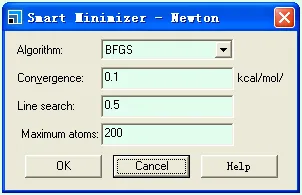
Smart Minimizer中,牛顿法可以设定最大的原子数,如果体系的原子数大于所设定的值,则计算是会自动地转为前面设定的收敛法(共轭梯度法或最陡下降法),收敛精度会改为共轭梯度法的默认收敛精度值。
Au(111)表面磺酸甜菜碱封端封端癸烷基硫醇自组装单层膜MS结构优化过程
盒子尺寸:34.609×29.970×91.91874Å
水层:20 Å,水密度为1g/cm3
真空层:40 Å(以避免镜像重叠,更好地模拟表面)
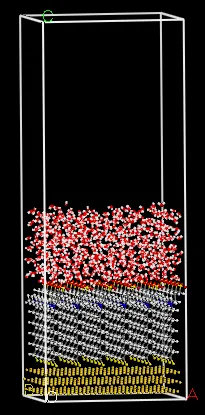
分子力学模拟设置
分子力场采用COMPASS,截断半径为12.5 Ǻ,计算范德华作用和库仑力作用的加和方法采用Atom Based,迭代方法采用Smart Minimizer。
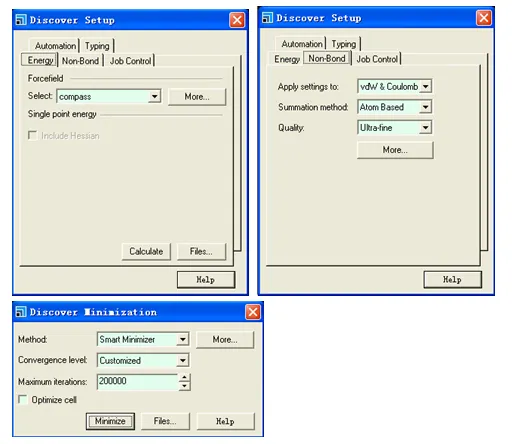
计算结果

免责声明:本文系网络转载或改编,未找到原创作者,版权归原作者所有。如涉及版权,请联系删
武汉格发信息技术有限公司,格发许可优化管理系统可以帮你评估贵公司软件许可的真实需求,再低成本合规性管理软件许可,帮助贵司提高软件投资回报率,为软件采购、使用提供科学决策依据。支持的软件有: CAD,CAE,PDM,PLM,Catia,Ugnx, AutoCAD, Pro/E, Solidworks 等。
 技术文档
技术文档
 推荐好文
推荐好文






 155-2731-8020
155-2731-8020




















