















































软件

产品



BIOVIA Materials Studio 2020是BIOVIA的化学和材料科学研究预测科学工具的最新版本。Materials Studio使研究人员能够理解材料的分子或晶体结构与其性质之间的关系,以便在材料研发方面做出更明智的决策。
通常,材料性能受多尺度现象影响。使用Materials Studio 2020的科学家拥有一套广泛的世界级求解器和参数集,可以从原子到微观尺度进行操作,模拟比以往任何时候都多的材料和属性。此外,现在可以在多物理建模和系统建模软件(如SIMULIA Abaqus和CATIA Dymola)中使用预测的属性来预测宏观行为。通过这种方式,可以使用多尺度模拟来解决材料设计和产品优化中一些最棘手的问题。
更好的材料 - 更好的电池
安全,快速充电,高能量密度和长寿命的电池迫切需要用于许多应用 - 尤其是作为化石燃料能源替代品的所有交通方式的电气化。电池设计依赖于从材料的最小尺度(电子结构)到电池单元和包装设计的几何形状的热力学,机械和化学过程之间的复杂相互作用。
改进用于电池和电容器的组件材料对于提供所需的性能提升至关重要。
Materials Studio提供了新功能,以实现液体电解质和电极组件的关键材料参数的模拟。

图1 原子电池模型的图像,显示电极、电解质和 SEI 层
1.新协议! Materials Studio Collection中添加了一个新的“质量和电荷传输”协议,以允许计算复杂电解液溶液的质量和电荷传输特性。它专门为液体电池电解质配方配置,并返回运行系统级别电池单元模型所需的锂离子传输特性,例如由CATIA Dymola [1]提供的模型。现在可以使用最终电池单元性能作为其中一个标准来优化电解质配方。
2.新协议! Materials Studio Collection中添加了第二个协议,用于计算离子插入电极结构产生的开路电压。这是配置为使用锂插入候选阳极材料(例如C,Si,Ge)或复杂阴极材料的稳定模型,但可以使用任何离子。在多组分阴极材料的情况下,可以使用由集群扩展方法生成的结构(也可在Materials Studio Collection中使用)进行准确表示。“开放式电池电压”协议返回了使用所提供离子的材料的半单元势差特性。
该协议默认使用CASTEP,但可以在任何提供总能量的服务器上运行。
金属合金 - 从密度泛函理论到CALPHAD数据库
金属合金材料的结构特征是由其所含的不同稳定原子晶体相产生的复杂晶粒结构。这些相反映了作为组成,温度和压力函数出现的最低能量配置,可以使用基于密度泛函导出能量的簇扩展和相关方法进行预测。这些方法解决了准确表示复杂金属混合物的组合问题。
BIOVIA Materials Studio Collection已经包含了一套方便且广泛的工具来生成这些结构,基于ATAT工具[1]。2020版本的新功能进一步扩展了这一点,使用稳定晶体结构集和液相模拟来生成TDB格式的CALPHAD数据库。亚稳相也可以包括在内,以提高应包括的相的覆盖范围。
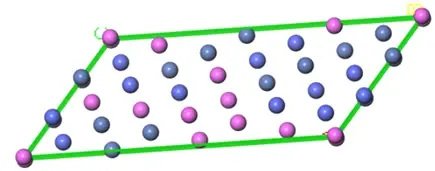
图2 特殊准随机结构 (SQS) 的示例 - 通常是 CALPHAD 数据库创建过程的一部分
CALPHAD(计算机耦合相图和热化学)数据库提供了一种方便的方式来捕获多组分相空间中的吉布斯自由能,并使用Pandat [2],ThermoCalc [3]和FactSage [4]等程序绘制相图。
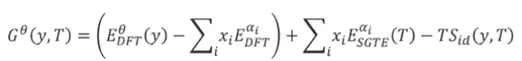
图3 吉布斯自由能的表达式作为模型相的函数, - 独立拟合用于填充 CALPHAD 数据库的 SQS 模型的 DFT 总能量
使用BIOVIA Materials Studio Collection中强大的金属合金工具包,现在可以探索预期在固化过程中出现在硅设计合金材料中的相。
TBD格式文件也可以用于为Phase Field模拟代码提供输入,以准确地表示晶粒和枝晶本身的发展。因此,CALPHAD数据库是原子尺度和介观尺度之间的重要桥梁技术,适用于这类材料。
更多参数 - 更好的科学!
经典模拟 使用基于经典力场的模拟进行预测的可靠性取决于描述构成原子或珠子之间相互作用的参数。在参数没有明确拟合,或者在最坏的情况下完全缺失的情况下,结果会受到严重影响。在BIOVIA Materials Studio Collection 2020中,现在可以使用协议进行新参数的自定义拟合。
新协议!对于价态参数(键伸展,角度弯曲和扭转),通过DMol3确定的能量和力的自动拟合被编码到一个Pipeline Pilot协议中。
新协议!第二个配置为将范德华参数拟合到凝聚相材料性质。
包含详细的教程,以便逐步介绍添加新参数的示例。可以通过Pipeline Pilot Connector直接从Materials Studio启动参数化。
新参数!BIOVIA Materials Studio COMPASS[5]一直被认为是支持多类材料的经典模拟的世界领先力场。
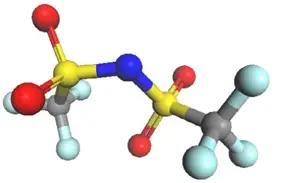
图4 双[(三氟甲基)磺酰基]亚胺 – 这种离子液体的锂盐,一种流行的锂离子电池电解质,已针对 COMPASS III 进行了改装
Materials Studio 2020标志着这些参数的下一代,COMPASS III[6],利用自动拟合程序并改进了离子液体支持。COMPASS III是将Maybridge[7],PoLyInfo [8]和ILThermo [9]数据库中发现的数千种常见结构进行拟合的结果,以产生广泛材料的高质量参数。
量子力学 - DFT泛函
新的CASTEP泛函!RSCAN Meta-GGA泛函[10]已被添加到BIOVIA Materials Studio CASTEP。
这是最近的通用SCAN泛函[11]的规范化版本,消除了原始实现中发现的一些不稳定性。RSCAN适用于广泛的固态和分子系统,准确度高。它可以用于使用有限位移法进行声子计算以及NMR性质(J-耦合除外)。
更多的DMol3泛函!在DMol3中,基于实现标准libXC库,已经公开了额外的混合泛函。现在可用的选项包括PBE0,TPSSh,SCAN0,M06和M06-2X。它们可用于执行QSAR,QMERA和Materials Studio Collection中的DMol3。
DFTB+参数化
新脚本!在Materials Studio安装的Examples/Scripting文件夹中提供了自动化DFTB+参数化程序的脚本。这些脚本大大简化了为一组元素对创建Slater-Koster文件的电子部分和排斥部分的过程。有一个教程“为DFTB+创建参数”可用来详细描述所需步骤。
COSMO-RS COSMOBase
新功能!已添加一个选项,用于以与DMol3-PBE COSMOBase一致的设置运行DMol3 COSMO。现在可以使用Materials Studio创建COSMO文件,并将它们转移到COSMOtherm[12]中使用。
BIOVIA COSMOtherm实现了COSMO-RS理论,用于精确计算纯流体和液体混合物的溶解度,活性系数,相图,蒸气压,汽化热和相平衡。
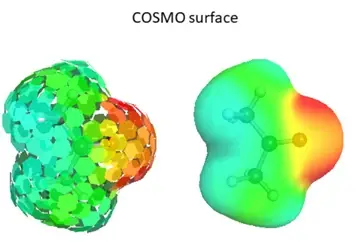
图5 COSMO 表面表示
大型金属系统
新功能!BIOVIA Materials Studio ONETEP已经扩展,提供了大型金属系统的线性缩放建模。已经成功地将退火和淬火算法用于费米算子展开(AQUA-FOE)方法[13]应用于在数千个核心上建模大型纳米簇模型,以开发新的催化剂材料[14]。
性能和可用性
新功能!已将多时间步长方法添加到Forcite中,现在是使用Ewald和PPPM静电力进行计算的默认方法。每四个时间步长就进行一次静电计算的倒空间部分,这对计算能量的准确性几乎没有影响,但对于多处理器计算可以显著提高性能。
新功能!在Materials Studio中通过Pipeline Pilot Connector运行计算时,您现在可以重用以前协议的设置来填充输入字段。
新协议!已将组件添加到Materials Studio Collection中,以增加直接从BIOVIA Materials Studio运行LAMMPS [15]分子动力学的能力。还提供了使用此组件的示例协议。
属性和分析
COOP/COHP分析
新功能!已将Crystal Orbital Overlap和Crystal Orbital Hamiltonian Population分析添加到DMol3。
这使得可以分析分子晶体中的键合。COOP/COHP可以作为通过轨道能量键合强度的指标,并且可以用于将DOS解析为键合/反键合态密度。
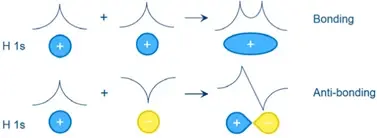
图6:化学家对分子轨道的键合/反键合性质感兴趣,COOP/COHP分析使得对晶体系统进行同样的洞察成为可能
Forcite和Mesocite
• 新功能!浓度剖面分析现在除了标准浓度剖面外,还以g/cm3的质量密度输出剖面。
• 新功能!当有足够的统计数据时,轨迹分析现在也通过自动拟合到均方位移数据直接计算扩散系数。
• 新功能!Forcite动力学现在通过MaterialsScript API支持振荡电场。
新脚本!现在有一个Materials Studio脚本可用,该脚本实现了所谓的Z-method [16],这是一种估计晶体模型(特别是金属)在不同压力下的熔化温度的方法。该脚本可以在Materials Studio安装的Examples/ Scripting文件夹中找到。
其他Materials Studio 2020亮点
CASTEP
• CASTEP用户界面已经进行了显著修改,以反映服务器代码的当前状态并提高可用性。
• 当请求NMR属性中的G-tensor时,现在使用EPR任务,以便在G-tensor计算中产生超精细张量。对超精细张量的计算文档已经更加明确。
• 现在可以从CASTEP分析对话框上的IR光谱选择访问振动分析工具。
• 为高通量研究生成的飞行势库(库QC5)已在Materials Studio脚本中提供。
• 包括工作函数计算在内的电静电势的可视化现在在CASTEP中可用于混合交换相关势。
GULP
• 在GULP中添加了模拟退火任务,以提供对分子动力学模拟中温度的灵活控制。
• GULP服务器代码已更新到学术版本5.2,其中主要的功能变化是:
- 允许在MD中使用多个温度坡度进行模拟退火。
- 偶极极化率(在非相互作用偶极子的极限下)现在具有二阶导数。
- 对于一般单胞而不仅仅是正交和单斜,允许进行领域分解。
- 添加了有限应变导数。
- 将自旋添加为物种属性,以支持Ising模型以及J2和J3耦合。
- 为MEAM添加了表面能量计算。
- 在ReaxFF中添加了孤对和过配位能量的改进平滑。
- 包括添加剪切力的选项。
- 添加了弹性常数的特征输出。
- Baskes势现在在二体组分中具有锥形。
- MEAM-2nn-QEq库扩展以包括Li-Co-O和Ti-O/Si-O系统的参数。
ONETEP
• ONETEP用户界面现在支持与导电态优化结合的局部DOS分析。
• 现在在ONETEP中实现了具有周期性边界条件的溶剂模型,以支持在溶剂中对体系和表面系统进行建模。
• 现在在ONETEP中提供了电子定位函数分析,作为可视化大系统中键合性质的一种方式。
• 现在可以使用PAW势进行ONETEP中电子激发的时间依赖DFT计算,除了之前可用的使用保守赝势的选项。
QMERA
• 已将使用DMol3计算拉曼光谱的功能添加到QMERA。
QSAR
• QSAR中的VAMP静电模型现在可以用于计算零微分重叠近似(ZDO)中的偶极矩。这是除了之前可用的自然原子轨道或点电荷(NAO/PC)近似之外的选项。
教程
已添加以下新教程:
新!使用Pipeline Pilot拟合价态力场参数:演示如何从Materials Studio运行Materials Studio Collection for Pipeline Pilot中的Fit Valence Parameters协议。
新!使用Pipeline Pilot拟合非键力场项:演示如何从Materials Studio运行Materials Studio Collection for Pipeline Pilot中的Fit VDW Parameters协议。
更新!DFTB+:为DFTB+创建参数:添加了一个关于确定良好电子设置的新部分。
还有许多教程已经使用2020功能,例如COMPASS III参数。
• DMol3:Diels-Alder反应的动力学
• Forcite:计算聚合物中气体的扩散率
• Pipeline Pilot Connector:使用Pipeline Pilot交联聚合物
• Pipeline Pilot Connector:使用Pipeline Pilot计算应力-应变图
• Reflex:使用近接接触惩罚解决4-硝基苯基己基脲的结构。
参考文献
1. https://www.brown.edu/Departments/Engineering/ Labs/avdw/atat/
2. http://www.computherm.com
3. https://www.thermocalc.com/
4. http://www.factsage.com/
5. COMPASS
6. Paper in Preparation.
7. https://www.maybridge.com
8. https://polymer.nims.go.jp/
9. https://ilthermo.boulder.nist.gov/
10. Bartók, A. P.; Yates, J. R. “Regularized SCAN functional”,J. Chem. Phys. ,150, 161101 (2019).
11. Sun, J.; Remsing, R. C.; Zhang, Y. ; Sun, Z.; Ruzsinszky, A.; Peng ,H.; Yang, Z. ; Paul, A. ; Waghmare, U. ;Wu, X. ;Klein, M. L. ; Perdew, J. P. Nat. Chem. 8 , 831 (2016).
12. http://www.cosmologic.de/
13. Aarons, J.; Skylaris, C.-K. “Electronic annealing Fermi operator expansion for DFT calculations on metallic systems”, J. Chem. Phys., 148, 074107 (2018).
14. L. G. Verga, J. Aarons, M. Sarwar, D. Thompsett, A. E. Russell, and C.-K. Skylaris, Faraday Discuss 208, 497 (2018) 15. https://lammps.sandia.gov/ 16. J. Phys.: Conf. Series 720 (2016) 012032

武汉格发信息技术有限公司,格发许可优化管理系统可以帮你评估贵公司软件许可的真实需求,再低成本合规性管理软件许可,帮助贵司提高软件投资回报率,为软件采购、使用提供科学决策依据。支持的软件有: CAD,CAE,PDM,PLM,Catia,Ugnx, AutoCAD, Pro/E, Solidworks 等。
 推荐好文
推荐好文






 155-2731-8020
155-2731-8020




















 技术文档
技术文档
